Our laboratory is currently working on three main projects: understanding antiviral silencing in plants, exploring the genomic variation of plant viruses, and determining the mechanisms of maize lethal necrosis disease. Below, you will find a brief overview of each project.
Project: Antiviral silencing in plants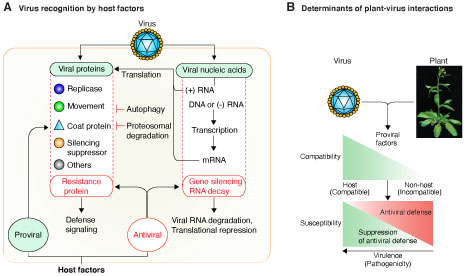 Viruses are identified as invasive by antiviral defense responses such as autophagy, ubiquitination, mRNA decay, and gene silencing, that target viral proteins or nucleic acids (Garcia-Ruiz, et al., 2019). In plants, gene silencing has a critical role in antiviral defense. Viruses are inducers and suppressors of RNA silencing. Thus, understanding the RNA silencing has potential to provide new insights into plant defense responses and in the mechanisms used by viruses to cause disease (Garcia-Ruiz, et al., 2018). The goal of this project is to determine the mechanisms of antiviral gene silencing, and has two parts: a) Identification viral RNA as invasive by the host into silencing machinery, and b) evasion, suppression or modulation of the host defense system by viruses to promote infection and cause disease. We have established genetically tractable model plant and model viruses. Multiple genetic analyses (Garcia-Ruiz, et al., 2015, and Garcia-Ruiz, et al., 2018) showed that establishment of virus infection is a two-step process determined initially by the availability of host factors needed for virus replication and movement. Subsequently, susceptibility is determined by the balance between host antiviral defense responses and suppression of defense by viral factors . This model provides the framework to identify triggers of antiviral defense in viruses of different genetic structure: positive strand RNA, negative strand RNA, or DNA.
|
Project: Maize lethal necrosis disease
Maize lethal necrosis disease is caused by the synergistic co-infection of MCMV and a member of the Potyviridae. Recently, we and other groups identified the poleroviruses Maize yellow mosaic virus (MaYMV), Maize yellow dwarf virus (MYDV-RMV), and Barley virus G in several parts of Africa (Wamaitha, et al., 2018). The molecular mechanisms of viral synergism in maize lethal necrosis remain to be determined. Our working model is that maize lethal necrosis is mediated by silencing suppressors encoded by the co-infecting viruses. These observations are consistent with a role for maize-infecting poleroviruses in maize lethal necrosis. For the first time, we have identified the silencing suppressor in MCMV, identified two silencing suppressors in SCMV, and we have determined that MYDV-RMV protein P0 is a strong silencing suppressor. Additionally, we have cloned MCMV, SCMV, MYDV-RMV, and have established a protocol to agro-infiltrate maize seedlings. These genetic resources will be used to determine the mechanisms of maize lethal necrosis disease and the role of poleroviruses as causal agents of maize lethal necrosis disease. |
Project: Genomic variation in plant viruses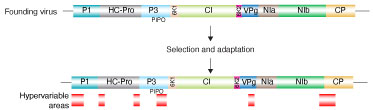 Potyviruses (family Potyviridae, genus Potyvirus) are transmitted in a non-persistent way by more than 200 species of aphids. As indicated by their wide host range, word-wide distribution, and diversity of their vectors, potyviruses have an outstanding capacity to adapt to new hosts and environments. Factors that confer adaptability are poorly understood. We hypothesized that selection imposed by hosts and vectors creates a footprint in areas of the genome involved in host adaptation. We profiled genomic and polyprotein variation in all species in the genus Potyvirus (Negam et. al. 2019) Results showed that the potyviral genome is under strong negative selection. Accordingly, the genome and polyprotein sequence are remarkably stable. However, nucleotide and amino acid substitutions across the potyviral genome are not randomly distributed and are not determined by codon usage. Instead, substitutions preferentially accumulate in hypervariable areas at homologous locations across potyviruses. At a frequency that is higher than that of the rest of the genome, hypervariable areas accumulate non-synonymous nucleotide substitutions and sites under positive selection. Our results show, for the first time, that there is correlation between host range and the frequency of sites under positive selection. Hypervariable areas map to the N terminal part of protein P1, N and C terminal parts of helper component proteinase (HC-Pro), the C terminal part of protein P3, VPg, the C terminal part of NIb (RNA-dependent RNA polymerase), and the N terminal part of the coat protein (CP). Additionally, a hypervariable area at the NIb-CP junction showed that there is variability in the sequence of the NIa protease cleavage sites. Structural alignment showed that the hypervariable area in the CP maps to the N terminal flexible loop and includes the motif required for aphid transmission. Collectively, results show that potyviruses contain fixed hypervariable areas in key parts of the genome which provide mutational robustness and are potentially involved in host adaptation. Hypervariable areas identified through computational methods provide the foundation for the biological characterization of viral, host and vector factors that mediate host adaptation in potyviruses. This part of the project allowed us to develop the computational framework to profile genomic variation in viruses using complete genome sequences and virus-derived siRNAs. This approach might be implemented to profile genomic variation in all kinds of viruses. We are currently profiling genomic variation in viruses of major importance in agriculture. |
